注意力缺陷多动障碍(ADHD)是儿童和青少年中最常见的神经发育障碍之一,主要表现为注意力不集中、多动和冲动行为,严重影响学习、社会功能及心理健康。尽管大量研究已揭示 ADHD 与额叶–纹状体环路功能异常及多巴胺信号紊乱密切相关,但越来越多的影像学和临床研究提示:海马结构与功能异常同样可能参与 ADHD 的发生发展。然而,海马脑区在 ADHD 发病机制中的具体作用及其分子基础,长期缺乏直接证据。整合素(integrins)是一类由 α、β 亚基组成的细胞黏附分子,广泛参与神经元迁移、树突发育、突触稳定及可塑性调控,是神经发育和突触成熟的重要分子基础。其中,整合素 α3 在兴奋性突触发育中的作用逐渐受到关注,但其功能异常是否与 ADHD 相关,尚不清楚。
近日,实验室认知神经科学中心肖晓研究员团队,联合复旦大学附属华山医院麻醉科王英伟教授团队,在 Molecular Psychiatry 期刊发表题为“Downregulation of Integrin α3 in ADHD mirrored in mutant mouse model by dopamine-dependent hippocampal AMPAR expression”的研究论文,首次系统揭示:前脑兴奋性神经元中整合素 α3 缺失可通过海马多巴胺 D1 受体依赖的 AMPAR 调控通路,引发 ADHD 样核心行为表型,并在小鼠模型与人类青少年队列中获得一致证据。
整合素α3缺失诱发性别差异性的
ADHD 样行为与海马结构异常
研究团队构建了前脑兴奋性神经元特异性整合素 α3 缺失小鼠(NEX-Itga3⁻/⁻),并在 P31 阶段系统评估其行为、脑结构及突触受体特征。结果显示:
1)行为学层面:雄性敲除小鼠表现出显著的多动、冲动、工作记忆受损及焦虑降低等 ADHD 样核心表型;雌性小鼠仅表现活动量增加,未出现明显 ADHD 样行为。
2)脑结构层面:敲除小鼠全脑体积减小,海马等皮层下结构体积显著缩小。
3)受体分布层面:皮层区谷氨酸受体分布未见异常,而海马 CA1 区突触后膜 AMPAR(GluA1)显著减少,NMDAR 上膜比例增加。
这些结果提示:整合素 α3 缺失主要影响海马突触成熟与功能,并伴随显著的性别差异性表型(图1)。
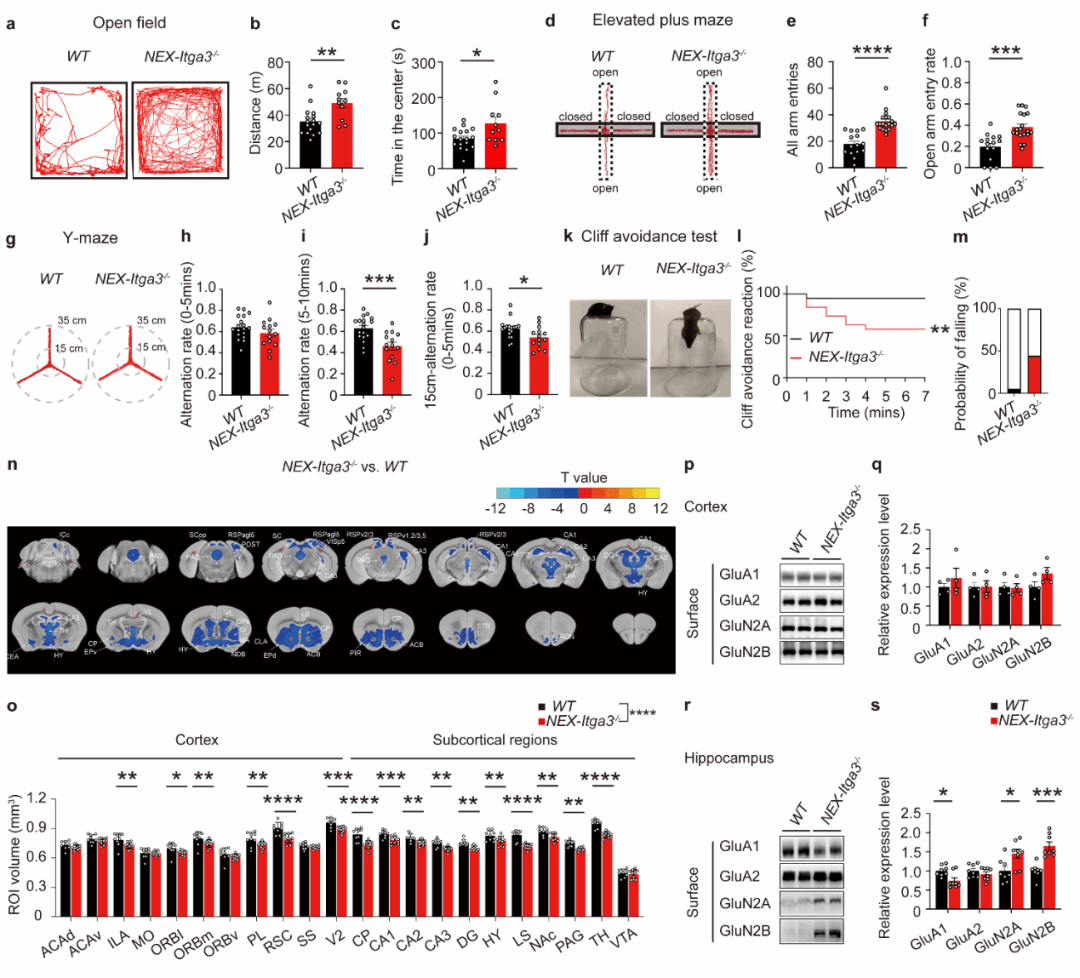
▲ 图1 Itgα3 缺失导致 ADHD 样行为,并伴随海马脑区体积缩小及 AMPAR 上膜减少
为进一步解析突触功能变化,研究团队在海马 CA1 锥体细胞中开展全细胞膜片钳记录与双光子谷氨酸解笼实验。结果发现:
1)AMPAR 介导的 mEPSCs 与 eEPSCs 幅度显著降低,变异系数(CV)升高,提示突触后效能下降、传递不稳定;
2)NMDAR 介导的 mEPSCs 未发生改变,但 eEPSCs 幅度升高;
3)双光子解笼实验显示 CA1 区存在大量 “沉默突触”,即仅表达 NMDAR、缺乏功能性 AMPAR 的未成熟突触。
上述结果表明,整合素 α3 缺失会阻碍海马突触由“未成熟”向“成熟”状态转变(图2)。
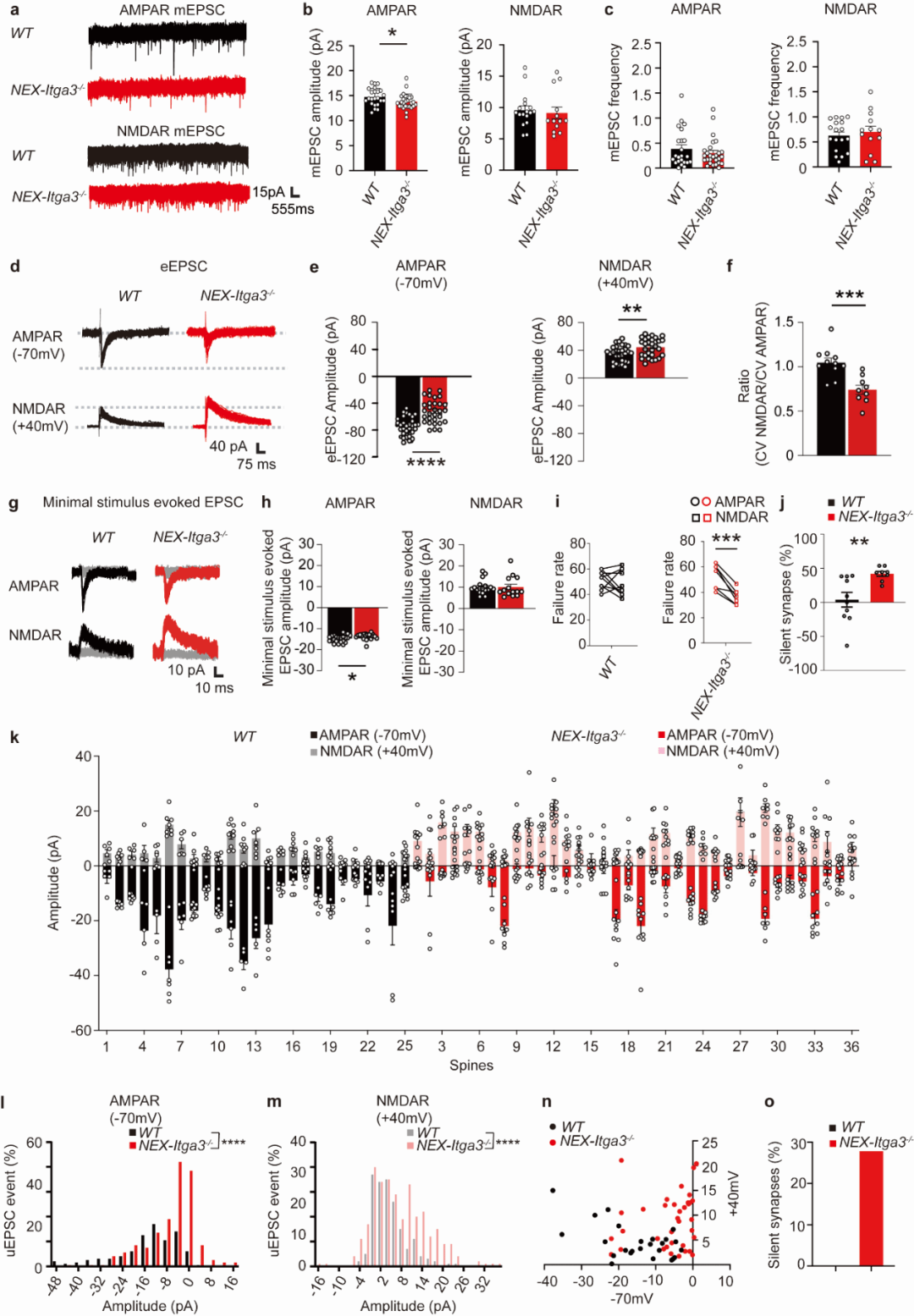
▲ 图2 Itgα3 缺失损害海马脑区突触后 AMPAR 功能并阻碍突触成熟
哌甲酯通过恢复海马AMPAR功能
挽救ADHD样行为
考虑到哌甲酯(MPH)是临床治疗 ADHD 的一线药物,研究团队对 NEX-Itga3⁻/⁻ 小鼠进行腹腔注射 MPH。行为学结果显示,MPH 可显著改善敲除小鼠的多动、冲动及注意力相关缺陷,在旷场、高架十字迷宫、Y 迷宫及避崖反应等多项测试中均表现出明显疗效(图3)。
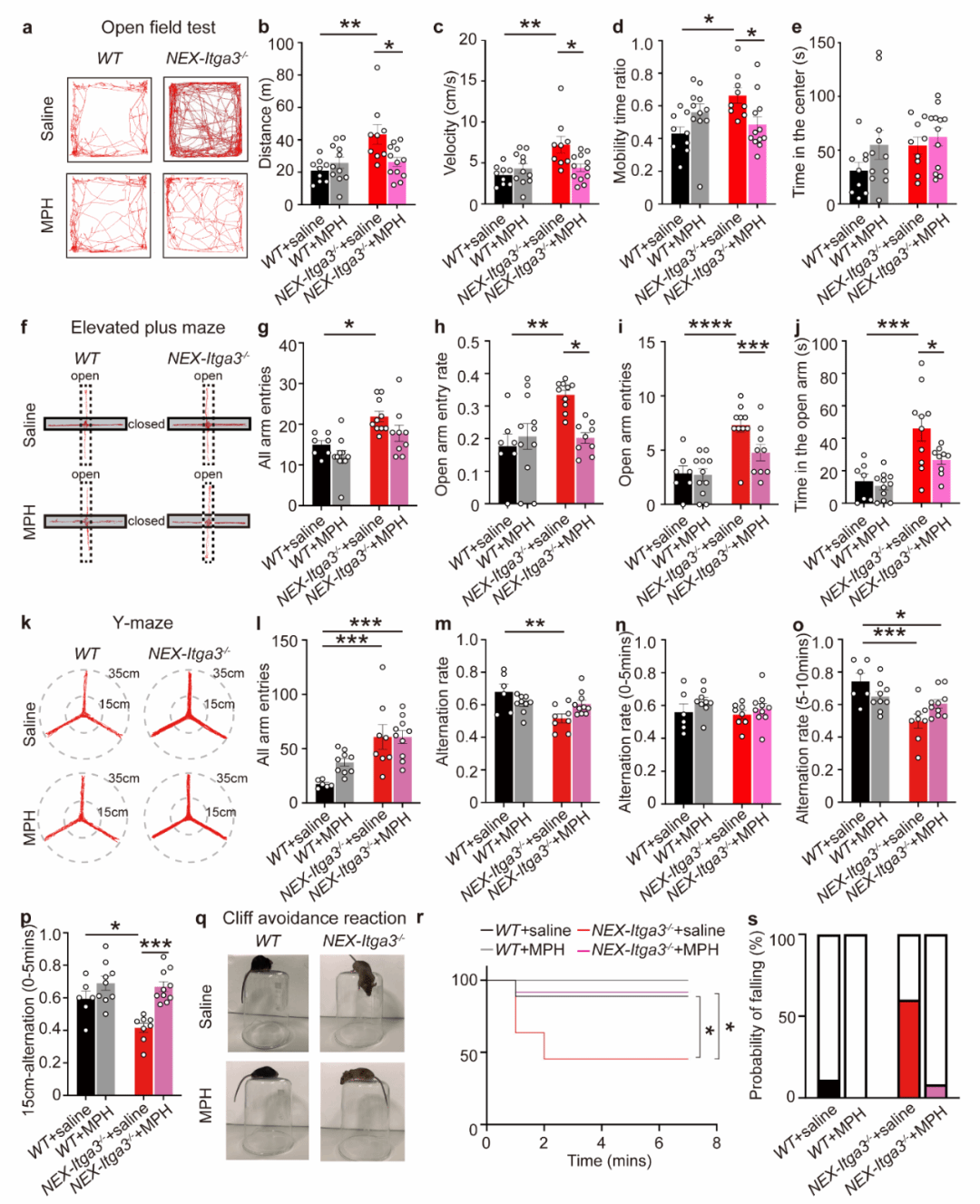
▲ 图3 哌甲酯改善NEX-Itgα3-/-小鼠的ADHD样行为
在分子与突触水平,MPH 能特异性恢复敲除小鼠海马 CA1 区 GluA1 的突触后膜表达,并同步增强 AMPAR 介导的突触电流。此外,研究发现敲除小鼠海马区多巴胺 D1 受体(Drd1)膜表面表达下降,而 MPH 处理可逆转这一改变(图4)。
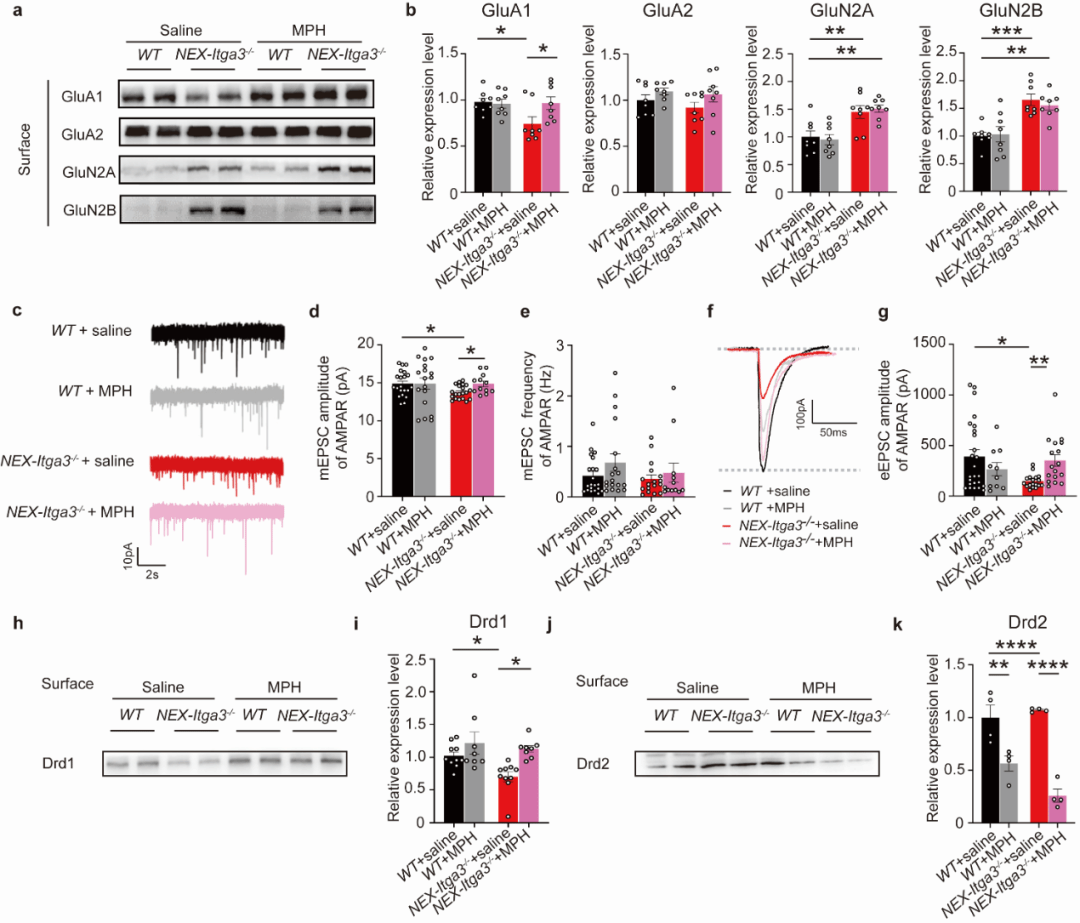
▲ 图4 MPH增加NEX-Itgα3-/-海马CA1区突触后膜AMPAR数量并恢复其功能
多巴胺 D1 而非 D2 受体特异性介导突触与行为修复
为进一步验证多巴胺受体亚型的作用,研究团队向敲除小鼠双侧背侧海马注射D1 受体激动剂 SKF81297 和D2 受体激动剂喹吡罗(QNP),结果显示:
1)SKF81297 可特异性恢复 AMPAR-mEPSC 幅度、GluA1 表面水平,并显著改善多动、冲动及认知缺陷;
2)QNP 虽可增强 AMPAR-mEPSC 幅度,但反而降低 AMPAR 表面表达,未能改善行为,甚至可能加重认知障碍。
这些结果明确表明:Drd1 依赖的 AMPAR 调控是整合素 α3 缺失导致ADHD 样表型的关键机制(图5、6)。
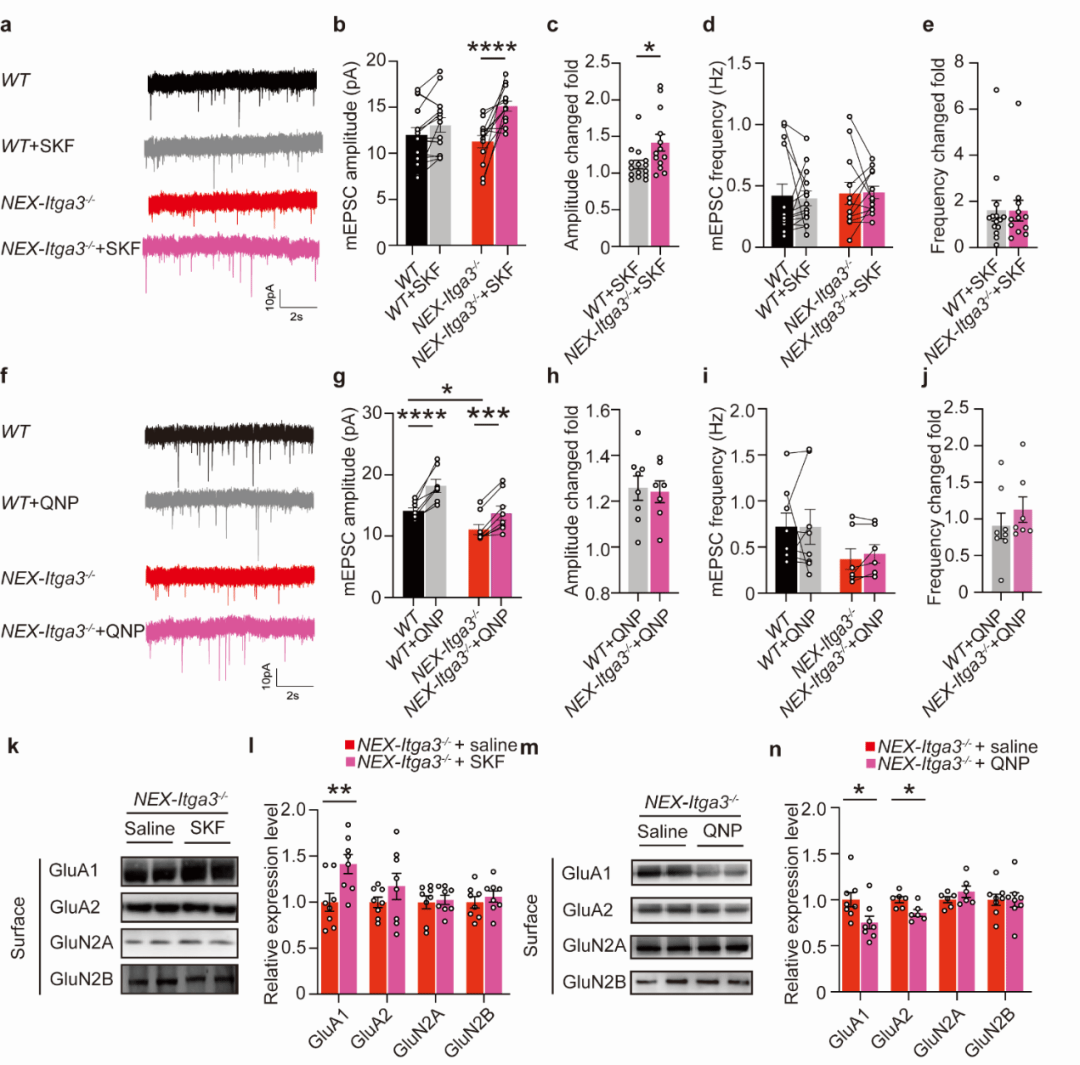
▲ 图5 SKF81297改善NEX-Itga3-/-小鼠AMPAR的电生理特性和突触后膜分布
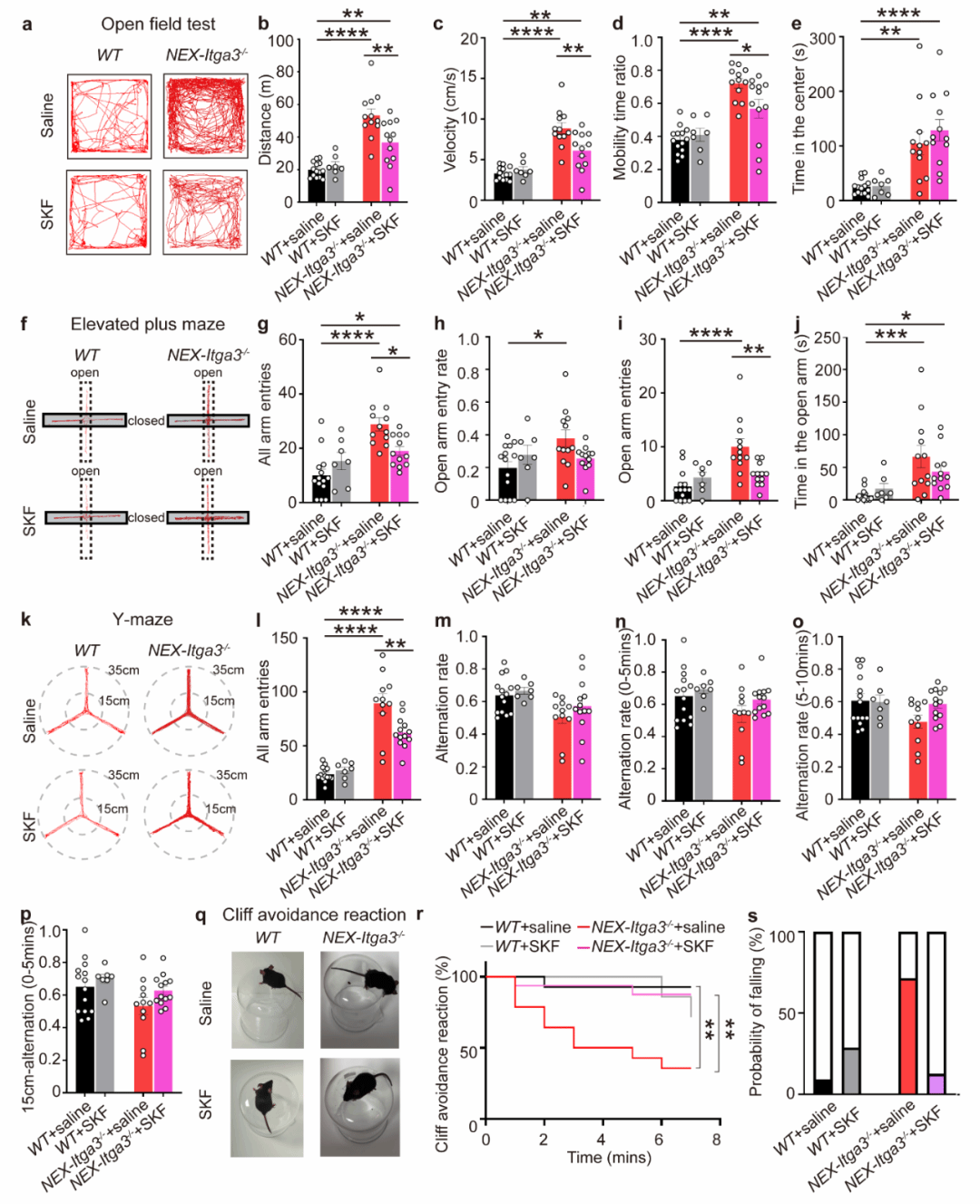
▲ 图6 SKF81297改善NEX-Itga3-/-小鼠ADHD样行为
人类队列研究证实 ITGA3 下调与男性 ADHD 风险相关
在跨物种验证层面,研究团队结合 GTEx 数据库与 ABCD(Adolescent Brain Cognitive Development)青少年队列,构建 ITGA3 加权多基因表达评分(PES)。分析发现:男性 ADHD 患者的 ITGA3-PES 显著高于对照组(提示 ITGA3 表达更低), ITGA3 表达降低与 ADHD 症状在基线及 4 年随访中均呈显著正相关,该关联在女性中未观察到;整合素 β1(ITGB1)呈类似关联,而其他整合素亚型未见显著关系。
该结果进一步支持整合素 α3β1 复合物在男性 ADHD 发病中的特异性作用(图7)。
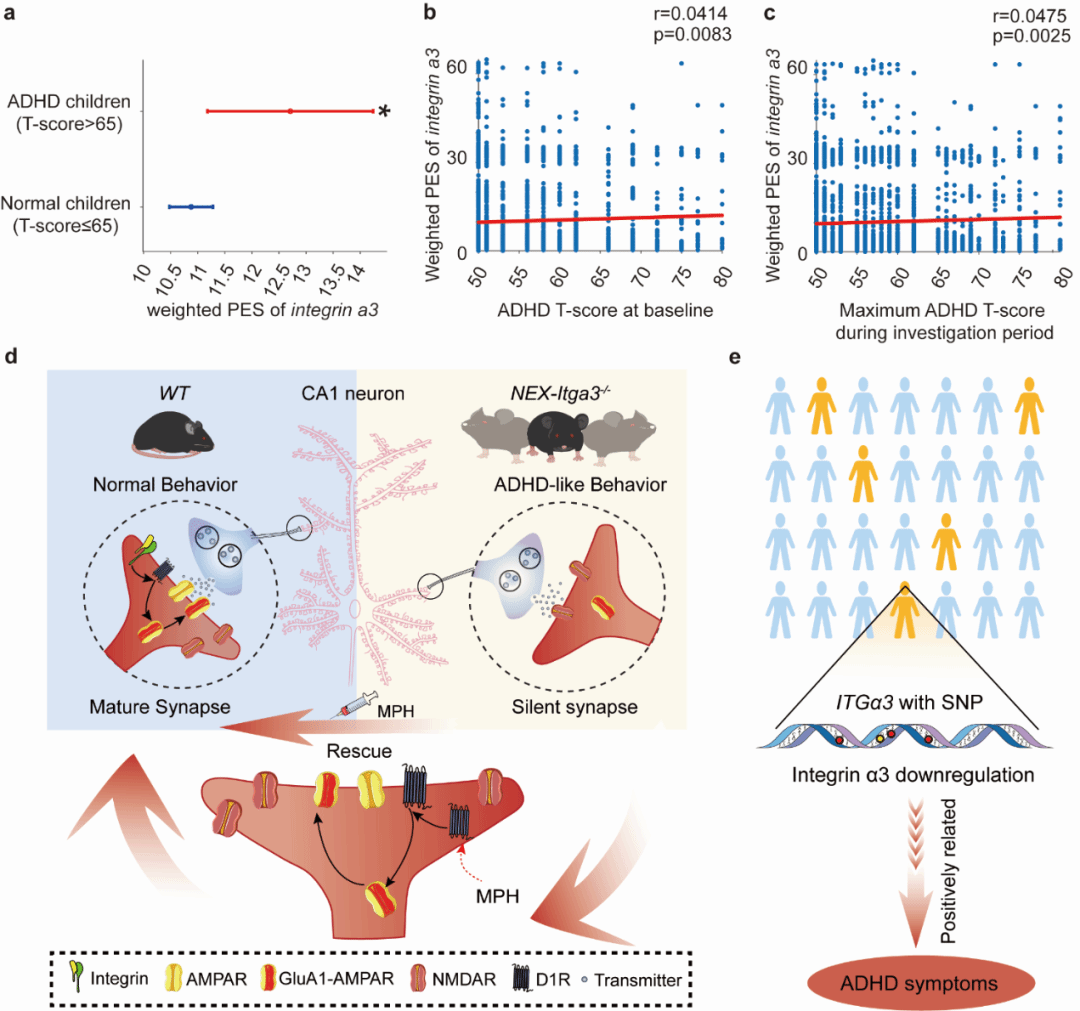
▲ 图7 整合素 α3 下调在小鼠模型和人类队列中均与 ADHD 显著相关
本研究首次从分子、突触、环路、行为及人类遗传学多个层面系统揭示:整合素 α3 功能缺失可通过“海马–多巴胺 D1–AMPAR”通路引发 ADHD 样核心表型,突破了传统以额叶–纹状体为中心的 ADHD 研究框架,明确了海马突触成熟障碍在 ADHD 中的关键作用。该发现不仅为理解 ADHD 的神经生物学机制提供了新视角,也为开发靶向突触可塑性与海马环路的精准干预策略提供了理论基础。
原文链接:https://www.nature.com/articles/s41380-025-03399-x


